
S2944
Anti-SMN antibody, Mouse monoclonal
clone 2B1, purified from hybridoma cell culture
Synonym(s):
Anti-Survival of Motor Neurons
About This Item
Recommended Products


biological source
mouse
Quality Level
conjugate
unconjugated
antibody form
purified from hybridoma cell culture
antibody product type
primary antibodies
clone
2B1, monoclonal
form
buffered aqueous solution
species reactivity
Xenopus, human, mouse
concentration
~2 mg/mL
technique(s)
microarray: suitable
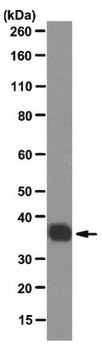
western blot: 2-4 μg/mL using A431 cell extract
isotype
IgG1
shipped in
dry ice
storage temp.
−20°C
target post-translational modification
unmodified
Gene Information
human ... SMN1(6606) , SMN2(6607)
mouse ... Smn1(20595)
General description
Specificity
Immunogen
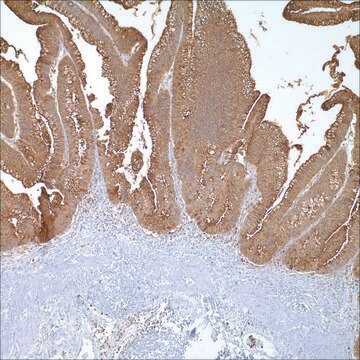
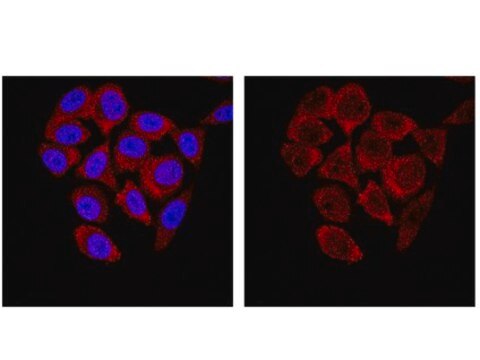
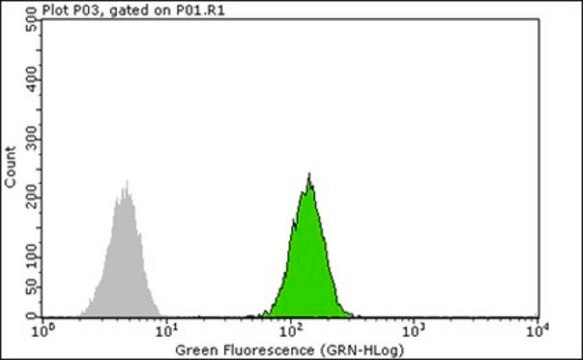
Application
Enzyme-linked immunosorbent assay (1 paper)
Biochem/physiol Actions
Physical form
Storage and Stability
Disclaimer
Not finding the right product?
Try our Product Selector Tool.
Storage Class Code
12 - Non Combustible Liquids
WGK
WGK 2
Flash Point(F)
Not applicable
Flash Point(C)
Not applicable
Certificates of Analysis (COA)
Search for Certificates of Analysis (COA) by entering the products Lot/Batch Number. Lot and Batch Numbers can be found on a product’s label following the words ‘Lot’ or ‘Batch’.
Already Own This Product?
Find documentation for the products that you have recently purchased in the Document Library.
Our team of scientists has experience in all areas of research including Life Science, Material Science, Chemical Synthesis, Chromatography, Analytical and many others.
Contact Technical Service